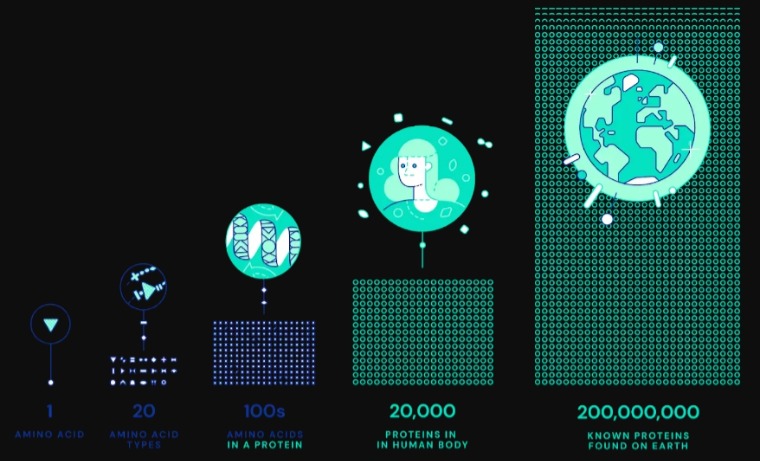
Proteins are complex molecules that allow our bodies to function. Protein induced misfolding, protein deficiencies and basically proteins not being able to carry out their functions are the root cause of all diseases. Proteins are made up of chains of 22 different amino acids which can come together to form trillion different combinations.
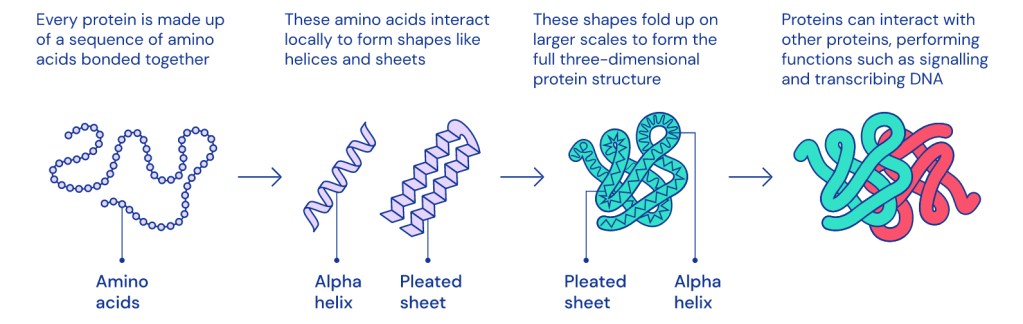
We know the amino acids at play, there chemical structure, and the shapes protein form (primary, secondary, tertiary, and quaternary). We know proteins fold into two primary structures first, alpha helices and beta sheets. From there these structures interact with each other to form more complex structures. We have analyzed 8 million sequences of all proteins (which is a growing number) and know of 20,000 proteins present in humans. With all of that, the question nagging scientists in molecular biology for the past 50 years, has been the “protein folding problem”.
The “protein folding problem” is the journey into understanding what shapes proteins to fold into its structure.
Source: DeepMind
The protein’s shape determines its function and being able to decode how a protein will fold, will open up the window to a new line of therapeutics.
When we unravel a protein it’s a string of amino acids and through attraction and repulsion between the amino acids, it causes the protein form its specific structure. The experimental techniques like nuclear magnetic resonance, X-ray crystallography and newer methods such as cryo-electron microscopy are expensive to carry out and take years to decode each protein structure.
SOURCE: DeepMind
The protein folding problem was first brought up in 1972 during an acceptance speech for Nobel Prize in Chemistry by Christian Anfinsen. Ever since scientists across the globe have been trying to crack this problem. It’s a difficult problem to crack because the number of conformations with the same string of amino acids can take on is astronomical. Cyrus Levinthal was a talented molecular biologist at MIT and lastly at Columbia. He estimated that there are 10300 possible conformations for a typical protein.
To tackle this problem more effectively in 1994 Professor John Moult and Professor Krzysztof Fidelis founded CASP. CASP stands for Critical Assessment of Protein Structure Prediction. In essence every two years the CASP hosts a community wide experiment were everyone is welcome to participate. They have had 14 so far since 1994. Before each CASP experiment, researchers all over the world submit a protein structures to decode. The proteins or region of a protein that CASP chooses are ones that have been recently experimentally determined. Then participants blindly predict the structure of the protein and these predictions are compared to the experimental data as it becomes available.
The metric used to measure the results of the CASP experiments is called GDT. It stands for Global Distance Test which has a range score of 0-100. It is the percentage of the type of amino acid residues/location within the threshold distance of the correct amino acids in the right location. In the latest CASP 14th assessment, from all the participants the median score was 92.4 GDT. A score of 90 or more gives enough confidence, that results from the blind assessment match up well with the experimental methods.
As mentioned earlier there have been 14 CASPs, and in CASP 13 something magical happened. Shown below in the figure provided by DeepMind, that in CASP 13 they saw the average GDT rise past 60% for the first time. Then in CASP 14 experiment the average GDT hits the magical 90%.
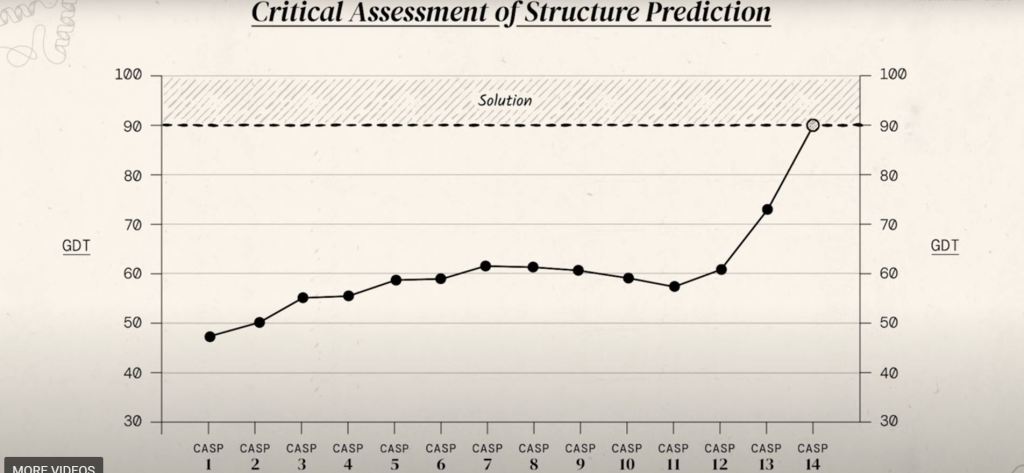
As experimental data and results were being published online for everyone to see, it started inspiring work being carried out by other scientists. Especially when DeepMind a research team in London, used their first iteration of AlphaFold in CASP 13.
AlphaFold is a deep learning architecture that can really accelerate protein structure discovery. The open source code has brought forward crucial implementation by scientists in the fields of structural biology, physics and bioinformatics.
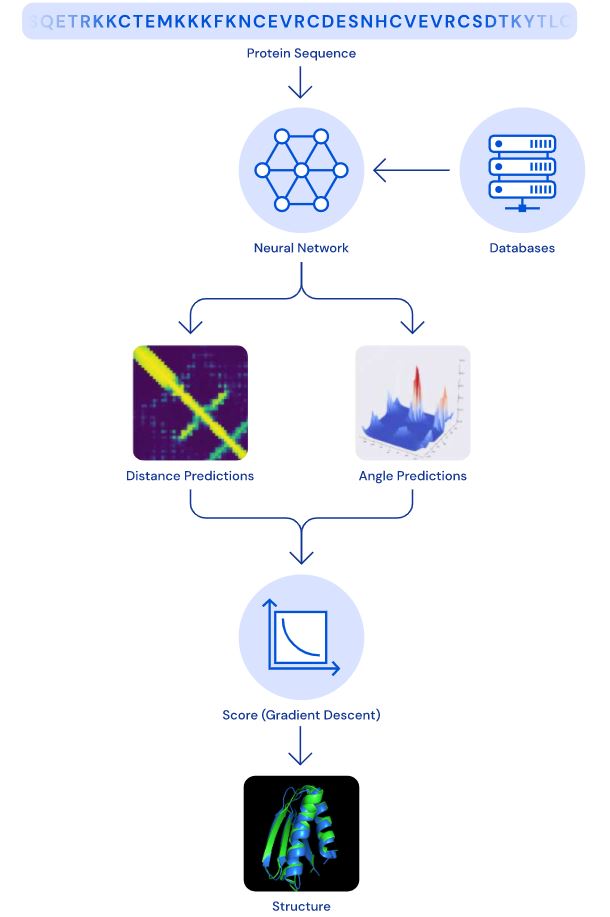
The two measurements that they were focusing on was (a) the distance that is between the pairs of amino acids and (b) the angles between chemical bonds that connect those amino acids.
SOURCE: DeepMind
The DeepMind team trained a neural network to predict the distance between every pair of residues in the protein. Using these probabilities AlphaFold can estimate the proposed protein structure. A separate neural structure uses the distance in aggregate to display how close the estimated structure is to the theoretical structure.
To then optimize the scores the team used a technique called a gradient descent. Which is a mathematical technique commonly used in machine learning to make precise improvements. All of this made CASP 13 possible.
Moving to the CASP 14 experiment Deep Mind’s expectations were higher to achieve a higher GDT score. Here they used their attention-based neural network which could effectively interpret the structure of the spatial graph developed by the folded protein. This allowed the team to use AlphaFold to predict which parts of the predicted protein structure can be trusted dependent on the internal confidence measure.
They trained the system using the publicly available protein data bank (170,000 protein structures), along with a large databank of protein sequences of unknown structure.

SOURCE: DeepMind
After hitting above 90% in GDT at CASP14 the team at DeepMind had truly achieved that vindication that Alpha Fold was here to stay. As said by former chairman and CEO of Genentech and current founder and CEO of Calico:
AlphaFold is a once in a generation advance, predicting protein structures with incredible speed and precision. This leap forward demonstrates how computational methods are poised to transform research in biology and hold much promise for accelerating the drug discovery process.
Arthur D. Levinson
AlphaFold as it becomes more robust, it will be a huge asset for drug discovery and finding new targets to create therapies. In CASP 14, Alpha Fold successfully predicted the SARS-CoV-2 viruses such as ORF3a whose structures were unknown. This can aid in rapidly developing vaccines for future pandemics.
There is a big but though. Even with Alpha Fold the power to move science rapidly is incredible but there are unfortunately a few caveats. Though there is a way to successfully predict an unknown protein structure, the biology of why the proteins form the complexes they do is rooted in the protein’s interactions of DNA, RNA and small molecules. The capacity to successfully pinpoint the location of all the amino acid side chains, is still not here. Looking back when CASP 1 started in 1994 and how far the program has come today, is something of an extraordinary feat. This should give us further confidence and excite us at what tools we will uncover just around the corner in the near future!
Citation
Senior, Andrew W, et al. “Improved Protein Structure Prediction Using Potentials from Deep Learning.” Springer Nature, 15 Jan. 2020, pp. 1–22.

Leave a comment